| Submission Procedure |
Dedicated Hardware for Biological Sequence Comparison
Abstract: Biological sequence comparison is a time consuming task on a Von Neuman computer. The addition of dedicated hardware for parallelizing the comparison algorithms results in a reduction of several orders of magnitude in the execution time. This paper presents and compares different dedicated approaches, based on the parallelization of the algorithms on linear arrays of processors. Key Words: Hardware, Biological Sequence Comparison, DNA, FPGA, VLSI Category: B.7.1 1 Introduction
Biological sequence comparison, such as the scanning of DNA or protein databases, is a fundamental task in molecular biology. This operation consists mainly of identifying sufficiently similar segments between two sequences. The computational complexity of this operation is proportional to the product of the length of the two sequences. Presently, software such as BLAST [1] or FASTA [10] are extensively used to perform the scanning of the biological databases. They have been designed to run on standard computers (i.e. Von Neuman machines) and include techniques for speeding up the process. These techniques are based on heuristics which can be tuned by setting external parameters. The search sensitivity depends mainly on these parameters. In general, a low sensitivity implies a short computation time (a few minutes), while a high sensitivity involves a very long computation time (a few hours). One could think that, in the future, the increasing power of the micro-processors would decrease the computation time. Unfortunately, the banks of sequences grow in size by approximately 50% per year and there is no reason to expect this progression to change in the next few years. Biological databases and micro-processor performance grow approximately at the same rate currently. As an example, the graph of the figure 1 shows two growing curves: the dark line represents the size of GenBank [3] (in million of nucleotides) since 1986; the dash line measures performance (in mips) of the fastest available 80x86 processor at introduction [8]. The two curves follow nearly the same exponential progression. Thus, biologists using Von Neuman computer will continue to have the dilemma of getting incomplete results in a short time or waiting a long time for satisfactory solutions. One way to limit the execution time is to parallelize the computation. Three approaches may be proposed to support task concurrency: computer networks, massively parallel machines or dedicated hardware. This paper focus only on the last category; it presents different dedicated hardware solutions which have been Page 77 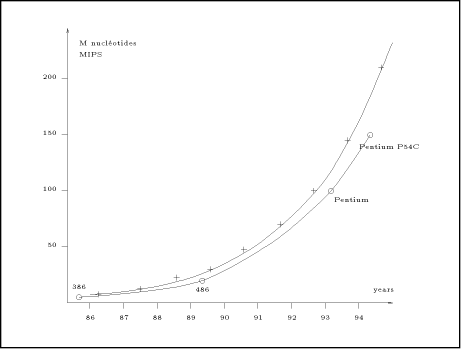
developed to speed up the sequence comparison process, and more precisely, the scanning of biological databases. We explain first how time-consuming algorithms, such as the Smith and Waterman algorithm, can be parallelized and implemented on a linear array of processors. Then different systems which have been fabricated and tested are briefly introduced and compared, before to conclude to some perspectives. 2 Basic Algorithm and Parallelization
Surprising relationships have been discovered between sequences that overall have little similarity. In that sense, the identification of similar segments (parts of the sequence) is probably the most useful and practical method for comparing two sequences. Fifteen years ago, Smith and Waterman [12] proposed a dynamic programming algorithm for detecting, between two sequences, the pair of segments which present high similarity. This algorithm compares two strings of characters by computing a distance which represents the minimal cost to transform one segment into another one by using two elementary operations: the substitution and the insertion/deletion operations. The last operation is called a gap operation. By using sequences of such operations any segment may be transform into any other segment. It is then possible to take the smallest number of operations Page 78 required to change one segment to another as the measure of distance between them. More formally, let X = ( H(i; j) is defined to be the maximum similarity of two segments ending
at A more complex formulation of the Smith and Waterman algorithm has
been proposed by Gotoh [6] to be closer to biological reality:
frequently, gaps of several adjacent bases are not the sum of single
gaps, but the result of one event. Thus, it is sometimes necessary to
weight these multiple gaps differently from summing single gap
weights: instead of having a single gap cost g, a cost function g(k)
is defined as g(k) = On the other hand, the Smith and Waterman algorithm can also be simplified by suppressing the gap penalty. In that case, only segments which have identical length are reported. Global alignment may also be found by omitting the zero maximum operation which prevents the H(i; j) value becoming negative and promotes the detection of local similarities. This algorithm can then be taken as a basis, knowing that many applications can be treated by applying slight modifications to the basic recursion. In the rest of the paper, and for the sake of clarity, we will consider only equation (1) as it constitutes mainly the bases for deriving the dedicated machine architectures. The idea employed to parallelize equation (1) is to associate one
processing element with each value H(i; j). Consider an array of n x m
processors denoted The overall operation of this two dimensional array is made on a
diagonal basis. Consider the comparison of two strings: assuming that
the computation starts at time 0, at time t = i + j + 1 all the
processors Since only one diagonal of this array is active at a time, the implementation may be done on a linear array. Each processor will have to perform the computation Page 79 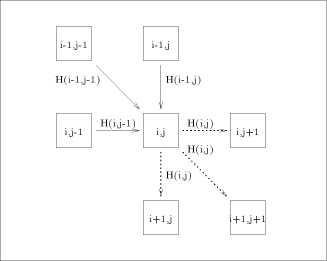
of either a row (n processors), a diagonal (n + m - 1 processors) or a column (m processors). The chosen projection may be directed by different criteria: - minimum number of processors; - maximum calculation throughput; - minimum data flow through the array. In the present case, two approaches are generally used for scanning biological databases. The choice depends on the algorithm: when segment match without gaps is desired, the diagonal emulation is preferred (figure 3-A); the query sequence and the sequences from the database are flowing through the array in opposite direction. For the rigorous Smith and Waterman algorithm, the col- umn emulation is used (figure 3-B). It consist of assigning each characters of the query sequence to one processor and feeding the array with the sequences of the database one character at a time. The speed up (
The speed up is expressed as the ratio of the sequential execution
time 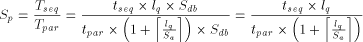 Page 80 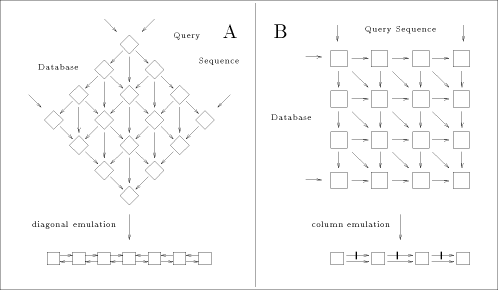
where 3 Dedicated Machines
Most of the machines which have been designed for speeding up the comparison of biological sequences are based on the linear array structures developed in the previous section. However, they differ in the flexibility of programming. We may distinguish three categories:
Page 81 Before comparing performances of these different machines, we first give a short overview of existing systems which have been specifically designed for molecular biological applications. This is not an exhaustive list: we took only the most representative and recent machines which have been fabricated and tested. 3.1 VLSI Dedicated arrays
BioScan [11] accelerates the identification of similar segments for DNA or protein sequences without allowing gap. It has been designed at the University of North Carolina. A chip contains 812 1-bit processors. A system with 16 chips is currently working, leading to a total number of 12,992 processors. In that implementation, scanning a database limits the query sequence to 12,992 characters. Presently, this is the most powerful system for detecting similar segments of identical length, but no commercial version is yet available. BISP (Biological Information Signal Processor) [5] provides a high speed and linearly-extensible system that can locate statistically similar subsequences of two DNA or protein sequences. It implements a modified version of the Smith and Waterman algorithm and allows many parameters to be set. The machine has been designed at the California Institute of technology. A chip contains 16 processors and a prototype machine of 16 chips has been validated, making possible the computation of unlimited sequence length. Again, this machine does not have a commercial version. SAMBA (Systolic Accelerator for Molecular Biological Application) is a machine which is very close to the BISP machine. A 128 processor version is currently running at IRISA. One chip contains only four processors which may be configured to compute local or global alignments with or without gap. It is an experimental machine which will be used locally for intensive bank to bank comparison. 3.2 FPGA arrays
Bioccelerator(1) is defined by its designer as an hybrid between application specific hardware and a general purpose computer. This is not a massively parallel machine since it includes only a maximum of 16 processors, but each processor can be configured to support many algorithms. This machine is marketed by Compugen LTD. SPLASH-2 [2] consists of a Sun SparcStation host, an interface board, and from one to sixteen Splash array boards containing each 16 FPGA processing elements. This system is not only dedicated to molecular biological application, even if the first version (Splash-1) has been mainly designed for that purpose. This machine is marketed under the WILDFIRE name by Annapolis Micro Systems, Inc. --------------(1) all the informations about the Bioccelerator machine has been taken from the following WEB server address http://sgbcd.weizmann.ac.il/BicMosaic.html Page 82 HScan [7] is 128 processor filter dedicated for scanning DNA databases. It has been developed at IRISA and validated on a FPGA platform, the PeRLe-1 prototype board [4]. It finds similar segments of identical length as BioScan does. The main difference between the other two systems is that it does not make exact calculation, but only detects the potentially interesting areas where similarities may appear. It is not yet a commercially available. 3.3 VLSI Programmable arrays
B-SYS [9] has be mainly designed for sequence comparison purpose, though is programming flexibility enables many other applications. This machine has been fabricated at the Brown University and tested on a 10 chip prototype leading to a total amount of 470 processors (47 processors per chip). 3.4 Performance comparison
There are many ways to compare the performances of dedicated machines. We will only consider the array peak performance, that is the maximum performance which can be reached, supposing that all the processors of the array are working and active. The measure unit is expressed in million of dynamic programming cell updates per second (MCUPS). A cell update represents the calculation of one H(i; j) of the equation (1). Table 1 shows the performances of the different systems we have briefly described. The MCUPS units must be considered with attention: BioScan and HScan perform identification of similar segments without allowing gap; this represents much less arithmetic and logic operations than the rigorous dynamic programming algorithm. The BioScan and the HScan systems are implemented on one single board
and have both been designed in the early 90s with comparable available
technology (CMOS 1.2 The BISP array can again be compared with the programmable B-SYS array
since they have both been designed in 1991, though with different
technologies: B-SYS has been integrated using a 2
Page 83 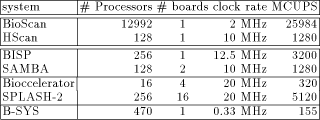 Table 1: This table gives the peak performances for different machines
dedicated to the biological sequence comparison in their maximum
configuration (column # Processors). The next column indicates how
many boards is required to support this configuration. Clock rate
represents the input frequency of the characters to the array; BioScan
chips are running a 32 MHz, but the 1-bit processors need 16 cycles to
perform a complete cell update. Similarly, the clock frequency of the
B-SYS chip is 10 MHz, but the programmable processors need about 30
cycles to compute a cell update. Finally, the MCUPS must be understood
as follows: the BioScan and HScan systems find segments without gaps
while the others system perform a complete Smith and Watermam
recursion.
Table 1: This table gives the peak performances for different machines
dedicated to the biological sequence comparison in their maximum
configuration (column # Processors). The next column indicates how
many boards is required to support this configuration. Clock rate
represents the input frequency of the characters to the array; BioScan
chips are running a 32 MHz, but the 1-bit processors need 16 cycles to
perform a complete cell update. Similarly, the clock frequency of the
B-SYS chip is 10 MHz, but the programmable processors need about 30
cycles to compute a cell update. Finally, the MCUPS must be understood
as follows: the BioScan and HScan systems find segments without gaps
while the others system perform a complete Smith and Watermam
recursion.
4 Conclusion
Until now, the exponential growth of biological sequence databases has been more or less compensated by the increasing power of micro-processors. There is no reason to expect changes in these two domains, though the biological database sizes seem to have a higher progression rate. However that may be, applications such as the scanning of biological databases will remain a time consuming task to determine weak similarities. Algorithms implied in the sequence comparison may be efficiently supported on parallel architectures. Today, a few systems have been proposed, fabricated and tested. They differ both on the programming flexibility and the hardware technologies. The performances of these systems may be resumed by the graph of the figure 4. Dedicated VLSI arrays are 20 times faster than FPGA or VLSI-programmable arrays, if we considered identical volumes of hardware having similar technology. FPGA and VLSI-programmable arrays may reach comparable performances, the FPGA low integration density/one clock cycle compensates the VLSI-programmable high integration density/several clock cycles. Another thing which is worth emphasizing is the availability of commercial machines. We may consider that Bioccelerator is the only commercial product, Page 84 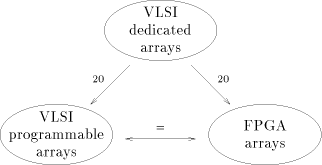 Figure 4: performances of the VLSI and FPGA systems. Dedicated VLSI arrays
are 20 times faster than FPGA or VLSI-programmable arrays. FPGA and VLSI-
programmable arrays have comparable performances
Figure 4: performances of the VLSI and FPGA systems. Dedicated VLSI arrays
are 20 times faster than FPGA or VLSI-programmable arrays. FPGA and VLSI-
programmable arrays have comparable performancessince SPLASH-2 (or WILDFIRE) is sold without biological software. Curiously, Bioccelerator is a FPGA machine with a very low level of parallelism (max of 16 processors). It takes advantage of the most recent FPGA technology while keeping off university approaches which advocate massive parallelism. Consequently, it does not offer the best performances compared to the other systems, but provides, although, an interesting speedup for a wide scale of biological applications. Tomorrow's VLSI technologies will provide on a single chip what we have today on a board. The question which maybe interesting to answer, right now, is: how can we anticipate the use of these future silicon resources for the comparison of biological sequences? Do we have to continue to fit into silicon very powerful arrays of processors targeted to some specific applications, or do we have to introduce more flexibility, leading to more modest performances? Looking at the present trends, the second solution seems to be more promising for a better future. References
[1] S.F. Altschul, W. Gish, W. Miller, E.W. Myers, and D.J. Lipman. Basic local alignment search tool. J. Biol. Mol, 215:403-410, 1990. [2] J.M. Arnold, D.A. Buell, and E.G. Davis. Splash-2. In 4th Annual ACM Symposium on Parallel Algorithms and Architectures, pages 316-322, 1992. [3] D. Benson, D. J. Lipman, and J. Ostell. Genbank. Nucl. Acids Res., 21(13):2963-2965, 1993. [4] P. Bertin, D. Roncin, and J. Vuillemin. Programmable active memories : a performance assessment. In F. Meyer auf der Heide, B. Monien, and A.L. Rosenberg, editors, Parallel Architectures and their efficient use, pages 119-130, Lecture notes in Computer Science, Springer-Verlag, Oct 1992. [5] E. Chow, T. Hunkapiller, and J. Peterson. Biological Information Signal Processor. In ASAP, pages 144-160, Sep 1991. [6] O. Gotoh. An Improved Algorith for Matching Biological Sequence. J. Mol. Biol., 162:705-708, 1982.
Page 85 [7] P. Guerdoux-Jamet and D. Lavenier. Systolic filter for fast dna similarity search. In ASAP'95, July 1995. [8] R. Halfhill. 80x86 wars. Byte, 74-88, June 1994. [9] R. P. Hughey. Programmable Systolic Arrays. PhD thesis, Brown University, May 1991. [10] W. R. Pearson and D.J. Lipman. Improved tools for biological sequence comparison. Proc. Natl. Acad. Sci., 85:3244-3248, 1988. [11] R.K. Singh, S.G. Tell, C.T. White, D. Hoffman, V.L. Chi, and B.W. Erickson. A Scalable Systolic Multiprocessor System for Analysis of Biological Sequences. In G. Borrielo and C. Ebeling, editors, Research on Integrated Systems, pages 168-182, 1993. [12] T.F. Smith and M.S. Waterman. Identification of common molecular subsequences. J. Mol. Biol, 147:195-197, 1981. Page 86
|
|||||||||||||||||
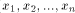 ) and Y = (
) and Y = (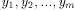 ) two sequences to be
compared. Let d(x; y) the substitution cost to change x into y and g
the cost of the insertion/deletion (gap) operation. The Smith and
Waterman algorithm is given by the following recursion:
) two sequences to be
compared. Let d(x; y) the substitution cost to change x into y and g
the cost of the insertion/deletion (gap) operation. The Smith and
Waterman algorithm is given by the following recursion: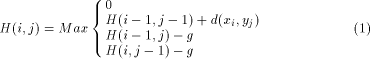 with H(i; 0) = H(0; j) = 0
with H(i; 0) = H(0; j) = 0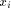 and
and 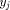 . From this point, a traceback procedure may be used to
determine the alignment between the two segments.
. From this point, a traceback procedure may be used to
determine the alignment between the two segments.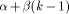 , where
, where  is the cost of the first gap and
is the cost of the first gap and 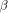 the cost
of the k - 1 following gaps. This improvement does not change the
recursion, the calculation of H(i; j) is just complicated.
the cost
of the k - 1 following gaps. This improvement does not change the
recursion, the calculation of H(i; j) is just complicated.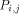 connected as indicated by the
connected as indicated by the 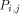 is able to perform the computation expressed by equation
(1).
is able to perform the computation expressed by equation
(1). 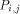 is represented by solid
arrows. H(i - 1, j - 1) is produced by
is represented by solid
arrows. H(i - 1, j - 1) is produced by 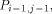 , H (i,j - 1) by
, H (i,j - 1) by 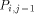 and H (i - 1,j) by
and H (i - 1,j) by 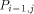 . With all this information, processor
. With all this information, processor 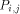 calculates H(i; j) and provides
the processors
calculates H(i; j) and provides
the processors 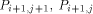 and
and 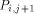 with the data they need. This data is represented
by dashed arrows on
with the data they need. This data is represented
by dashed arrows on 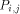 are active. In such a way, it can be easily checked that
all the arguments needed for the computation of equation (1) have
already been computed and have been routed correctly.
are active. In such a way, it can be easily checked that
all the arguments needed for the computation of equation (1) have
already been computed and have been routed correctly.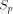 ) provided by these types of architectures - compared
to a sequential (Von Neuman) computer - comes from two levels of
parallelism:
) provided by these types of architectures - compared
to a sequential (Von Neuman) computer - comes from two levels of
parallelism: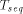 and the parallel execution time
and the parallel execution time 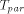 :
: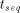 is the time for computing a recursion on a sequential computer,
is the time for computing a recursion on a sequential computer, 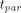 the time for computing the same recursion with dedicated hardware,
the time for computing the same recursion with dedicated hardware, 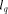 the
length of the query sequence,
the
length of the query sequence, 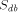 the size of the database and
the size of the database and 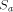 the size of
the array.
the size of
the array. 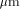 , for BioScan, and Xilinc 3090 for PeRLe-1);
however, the power difference is high (about 20). This comes
essentially from the technologies used: the VLSI technology is
generally about 2 or 3 time faster and about 10 times denser than the
FPGA technology. The same difference may be observed between the BISP
board and one SPLASH-2 board, by considering a technology up-to-date
of the BISP chip (designed and fabricated in 1991, 1
, for BioScan, and Xilinc 3090 for PeRLe-1);
however, the power difference is high (about 20). This comes
essentially from the technologies used: the VLSI technology is
generally about 2 or 3 time faster and about 10 times denser than the
FPGA technology. The same difference may be observed between the BISP
board and one SPLASH-2 board, by considering a technology up-to-date
of the BISP chip (designed and fabricated in 1991, 1 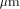 CMOS process)
compared to the actual Xilinx component (XC 4010) technology.
CMOS process)
compared to the actual Xilinx component (XC 4010) technology.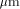 CMOS process. On the
other hand, the B-SYS processors (8-bit) are smaller than the BISP
processors (16-bit). An approximate power difference of 20 may be
established between the 2 approaches. If peak performances may give an
idea about the power capabilities of the three dedicated approaches,
these performances are never sustained in real systems. In fact, there
are two main reasons which limit the performances:
CMOS process. On the
other hand, the B-SYS processors (8-bit) are smaller than the BISP
processors (16-bit). An approximate power difference of 20 may be
established between the 2 approaches. If peak performances may give an
idea about the power capabilities of the three dedicated approaches,
these performances are never sustained in real systems. In fact, there
are two main reasons which limit the performances: